RMPとは医薬品リスク管理計画のことで、医薬品のリスクについて、開発・審査・市販後を通じて管理するための計画をまとめた資料になります。開発・審査の段階で明らかになっている副作用だけでは医薬品のリスクを確定するには不十分で、市販後に明らかになりうるものまでを想定して管理していくことが薬害を防ぐために重要です。
RMPとはRisk Management Planを略したもので、日本語で医薬品リスク管理計画と言います。
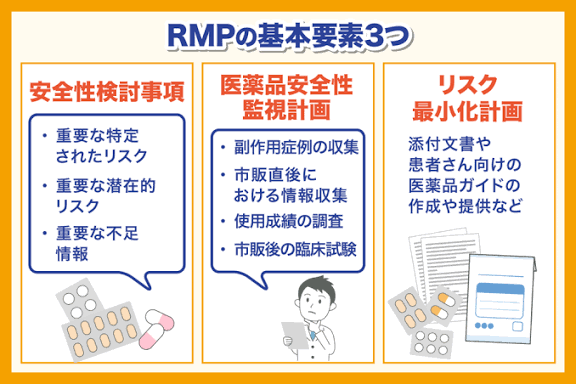
すべての医薬品にはリスク(副作用)がつきものです。
そのため、医薬品を開発する段階で非臨床試験や臨床試験が実施され、慎重に安全性のチェックが行われます。
ですが、市販後に使用される範囲に比べるとかなり限定的なものであることは否定できません。
開発段階で得られたデータは行政に提出され、有効性や安全性の評価に関する審査を受けます。
この段階で問題ないと判断されたものが承認され、医療用医薬品として販売されることになります。
市販開始後は開発段階よりも広い範囲で医薬品が使用されます。
高齢者や合併症のある患者、併用薬を服用中の患者など開発段階で十分に検証しきれていないようなケースに対して使用されていくことになります。
その結果、予期しないリスク(副作用)が発生したり、ベネフィット・リスクが損なわれる事象(副作用頻度の上昇、予想外の使用方法)が発生する可能性があります。
このように、医薬品の安全性を十分検証するためには、承認申請前の試験だけでは不十分で、市販後も継続して検証を行なっていくことが大切です。
そこで導入されたのが医薬品リスク管理計画(RMP)です。
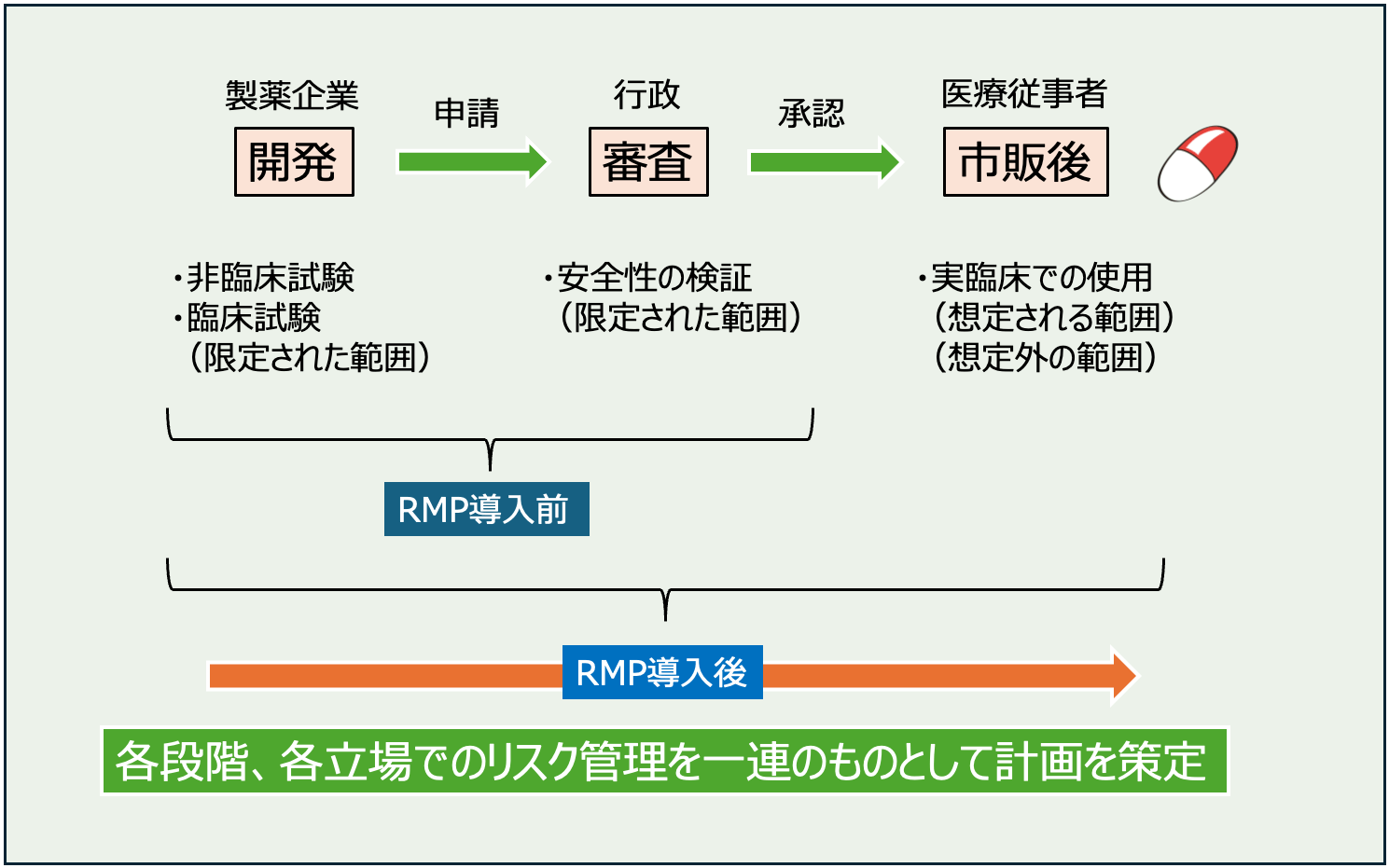
日本では2012年4月に「医薬品リスク管理計画指針」が公開され、2013年4月に正式に導入されました。
導入以降は新薬の承認時にRMPの案を提出し、承認審査の段階でリスク最小化計画が必要と判断されたものについては、RMPの策定が義務付けられ、添付文書等とともにPMDAのホームページで公開されます。
その場合、RMP策定と実施が医薬品の承認条件となり、そのことが添付文書の21.承認条件の項に記載されています。

ジャディアンスのRMPについて